При наличии заместителей в ядре бензола место вступления нитрогруппы и условия реакции определяются, как и при других реакциях электрофильного замещения природой имеющегося заместителя.
Ориентанты I рода (OH, OR, OCOR, NH 2 , NHR, NR 2 , NHCOR, –N=N–, CH 2 Cl, CH 3 , F, Cl, Br, I) направляют нитрогруппу преимущественно в орто-пара положения и облегчают (кроме галоидов) её вступление.
Ориентанты II рода (SO 3 H, NO 2 , COOH, COOR, CN, CCl 3) направляют нитрогруппу главным образом в мета-положение и затрудняют её введение в ядро.
Соединения, содержащие ориентанты I рода, нитруются в более мягких условиях: нитрование толуола до мононитросоединений происходит при температуре не выше 40°С; мононитрование фенола осуществляется даже разбавленной азотной кислотой при охлаждении. От характера имеющихся в ядре бензола заместителей зависит и скорость реакции. Сравнительное изменение скорости реакции под влиянием имеющегося заместителя схематически можно представить рядом, где группы, стоящие вправо от хлора, ускоряют реакцию, а влево – замедляют её.
NO 2 > SO 3 H > COOH > Cl < CH 3 < OCH 3 < OC 2 H 5 < OH
замедляют ускоряют
В случае ди- и полизамещенных бензолов влияние заместителей носит аддитивный характер. При наличии заместителей разного типа место вступления электрофила определяет ориентант I рода, так как он активирует ядро. Если оба заместителя одного рода, то место вступления определяет более сильный:
Нитрование толуола нитрующей смесью при 20-30°С приводит к получению смеси о- и n-нитротолуолов с почти количественным суммарным выходом.
Дальнейшее нитрование нитротолуолов до нитросоединений осуществляется при более высокой температуре.
 |
При действии дымящей азотной и серной кислот при 110°С на динитротолуолы образуется 2,4,6-тринитротолуол (тротил), применяющийся в качестве взрывчатого вещества. С увеличением числа алкильных групп в бензольном кольце нитрование облегчается. Ксилолы нитруются легче, чем толуол, а мезитилен в еще более мягких условиях ацетил- или бензоилнитратом.
Нитрование
Реакция нитрования является одной из наиболее изученных реакций ароматического замещения. Для препаративных целей нитрование, как правило, проводят смесью концентрированных азотной и серной кислот, так называемой нитрующей смесью . На первой стадии реакции происходит образование иона нитрония + NO 2 , который и является электрофильным агентом:
HO-NO 2 + H 2 SO 4 H 2 O + -NO 2 + HSO 4 -
H 2 O + -NO 2 + H 2 SO 4 H 3 O + + HSO 4 - + + NO 2
Наличие иона нитрония в этом растворе подтверждено спектроскопически. Азотная кислота в концентрированной серной кислоте практически нацело превращается в нитроний-катион. Незначительная эффективность самой азотной кислоты в реакции нитрования бензола объясняется низким содержанием иона + NO 2 .
B качестве нитрующих агентов используются также другие системы, в которых генерируется либо катион + NO 2 , либо соединение общей формулы NO2-Y где Y - хорошая уходящая группа. Некоторые из таких систем, нашедшие наибольшее применение, представлены в таблице 1 в порядке увеличения их активности.
Таблица 1. Нитрующие реагенты.
| Нитрующий реагент | Метод генерации | Арены, подвергающиеся нитрованию |
| Азотная кислота HO-NO2 |
Фенолы, эфиры фенолов, бифенил | |
| Ацетилнитрат CH 3 C(O)-O-NO 2 | СН 3 СООН + HNO 3 (СН 3 СО) 2 О + HNO 3 | Бензол, алкилбензолы |
| Диоксид азота N 2 O 4 (O=N-O-NO 2) |
Бензол, алкилбензолы | |
| Нитрующая смесь | H 2 SO 4 конц + HNO 3 | Бензол, алкилбензолы, галогенбензолы, бензойная кислота, нитробензол, нафталин |
| Хлорид нитрония Сl-NO 2 |
Бензол, алкилбензолы, нитробензол, | |
| Тетрафторборат нитрония BF 4 -+ NO 2 | HF . 2BF 3 + HNO 3 | Динитробензол |
На примере реакции нитрования алкилбензолов отчетливо прослеживается влияние пространственных факторов на направление электрофильного замещения. Так, при нитровании толуола (метилбензола) орто -изомер образуется в качестве основного продукта, а при переходе к этил-, изо -пропил- и особенно к трет -бутил-бензолу его выход существенно уменьшается (см. табл. 2).
Таблица 2. Влияние пространственых факторов на соотношение орто-, пара-изомеров в реакции нитрования (NO 2 +)
Замещение в положения, % |
|||
C 6 H 5 -C 2 H 5 |
|||
C 6 H 5 -CH(CH 3) 2 |
|||
C 6 H 5 -C(CH 3) 3 |
|||
При изучении нитрования алкилбензолов было обнаружено так называемое ипсо-замещение , когда электрофильная атака протекает по тому атому углерода бензольного кольца, которое уже содержит заместитель, например:

В отличие от нитрования, при галогенировании атака ароматического субстрата может осуществляться различными электрофилами. Свободные галогены, например, Cl 2 и Br 2 ,(прим.35) могут легко атаковать активированное ароматическое ядро (например, фенола), но не способны реагировать с бензолами и алкилбензолами (фотохимическая активация может, однако, в последнем случае привести к протеканию радикального замещения в боковую цепь; см. раздел IV.3). Для поляризации атакующей молекулы галогена необходим катализ кислотами Льюиса , такими как AlCl 3 , FeBr 3 , и т.п.; при этом в молекуле галогена появляется так называемый "электрофильный конец" (энергия же, требующаяся для образования катиона Наl + существенно выше). Тем самым электрофильное замещение существенно облегчается:

Галогенирование протекает очень энергично, если использовать реагенты, в которых галоген в результате поляризации имеет сильный положительный заряд или даже существует как катион. Так, очень инертный мета -динитробензол можно пробромировать бромом в концентрированной серной кислоте в присутствии сульфата серебра. Предполагают, что в этом случае промежуточно образуется бром-катион:
2Br 2 + Ag 2 SO 4 2Br + + 2AgBr + SO 4 2-
Реакционная способность элементарного иода в реакциях электрофильного замещения в ароматическом ядре незначительна, так что прямое иодирование возможно только в случае фенола и ароматических аминов. Иодирование других ароматических соединений проводят в присутствии окислителя (обычно, азотной кислоты). Считается, что в этих условиях роль электрофильного агента играет ион I- + OH 2 .

Для галогенирования аренов можно применять также смешанные галогены , например, монохлорид брома (BrCl) или иода (ICl):


Галогенирование in vivo . В качестве примера электрофильного ароматического галогенирования, протекающего в живых организмах, можно привести реакцию иодирования -аминокислоты - тирозина в ходе биосинтеза иодсодержащих гормонов щитовидной железы до 3-иодтирозина и далее до 3,5-дииодтирозина:

Детали механизма сульфирования исследованы менее подробно по сравнению с нитрованием и галогенированием. Сам бензол сульфируется довольно медленно горячей концентрированной серной кислотой, но быстро - олеумом, SO 3 в инертных растворителях или комплексом SO 3 с пиридином. Природа электрофильной частицы зависит от условий реакции, но, вероятно, это всегда SO 3 , или в свободном состоянии, или связанный с "носителем", например, в виде H 2 SO 4 . SO 3 (H 2 S 2 O 7) в серной кислоте. Небольшие количества SO 3 образуются в H 2 SO 4:
2H 2 SO 4 SO 3 + H 3 O + + HSO 4 -
Атаку ароматического субстрата осуществляет атом серы поскольку он сильно положительно поляризован, то есть электронодефицитен:

Сульфирование является обратимым процессом. Это имеет практическое значение: при обработке сульфокислот водяным паромпроисходит замещение группы SO 3 Н на водород. Таким образом, можно ввести группу SO 3 Н как заместитель, ориентирующий требуемым образом последующие реакции (см. раздел IV.1.Б), а затем ее отщепить. Некоторые интересные особенности имеет сульфирование нафталина (см. раздел IV.1.Г).
Подобно галогенам, алкилгалогениды могут быть так сильно поляризованы кислотами Льюиса (хлоридами алюминия и цинка, трифторидом бора и др.), что они становятся способными к электрофильному замещению в ароматическом ядре:
R-Cl + AlCl 3 R +... Cl ...- AlCl 3 R + AlCl 4 -
Кроме алкилгалогенидов, источниками карбокатионов для галогенирования ароматических соединений могут быть алкены или спирты. При этом необходимо присутствие протонной кислоты, чтобы протонировать алкен или спирт. В случае спиртов требуется добавка не менее чем эквимольного количества кислоты (так как вода, выделяющаяся в ходе реакции, дезактивирует эквимольное количество катализатора), тогда как в реакциях с участием алкилгалогенидов и алкенов достаточно добавлять незначительное количество катализатора.
В лаборатории алкилирование по Фриделю-Крафтсу имеет ограниченное применение, так как обычно при этой реакции образуются смеси продуктов, что обусловлено рядом причин:
1) Образующийся продукт алкилирования легче вступает в реакции электрофильного ароматического замещения, чем исходное соединение (Alk - электронодонорная группа), поэтому дальше преимущественно алкилируется продукт. Если хотят получить продукты моноалкилирования, то необходимо брать большой избыток ароматического соединения.
2) Как и сульфирование, реакция алкилирования по Фриделю-Крафтсу обратима (см. также раздел IV.1.Г).
3) Даже в мягких условиях первичные и вторичные алкилгалогениды дают преимущественно вторичные или третичные алкиларены соответственно, поскольку алкилирование происходит в условиях, приближающихся к S N 1 реакции.(прим.37) Перегруппировки можно избежать, если работать при низких температурах.
Ацилирование ароматических соединений по Фриделю-Крафтсу является важнейшим методом синтеза жирноароматических кетонов. Производные карбоновых кислот, такие как ацилгалогениды и ангидриды, имеют полярную карбонильную группу и в принципе способны к электрофильному замещению в ароматических системах:

Электрофильная активность этих соединений, однако, невелика, и должна быть повышена действием кислот Льюиса. При этом кислотный катализатор, как правило, атакует атом кислорода карбонильного соединения и, смещая электронную плотность, повышает положительный заряд соседнего атома углерода. В результате образуется поляризованный комплекс (а в пределе - ацилкатион), действующий как электрофил:

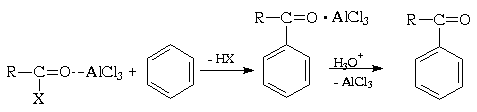
Важное отличие реакции ацилирования ацилгалогенидами от реакции алкилирования алкилгалогенидами состоит в том, что в первой из этих реакций требуется более 1 моль кислоты Льюиса , тогда как во второй необходимо только каталитические количество. Это обусловлено тем, что кислота Льюиса образует комплекс как с ацилирующим производным карбоновой кислоты, так и с кетоном - продуктом реакции. При взаимодействии с ангидридами получающаяся кислота связывает еще моль моль катализатора, так что в целом его необходимо по крайней мере два моль. В каждом случае по окончании реакции образовавшийся комплекс кетона с хлоридом алюминия (или другой кислотой Льюиса) должен быть гидролитически разрушен (соляной кислотой со льдом).
Полиацилирования не наблюдается, поскольку образующийся кетон значительно менее реакционноспособен, чем исходное соединение (см. раздел IV.1.Б). Поэтому алкилбензолы часто предпочитают получать не прямым алкилированием, а ацилированием по Фриделю-Крафтсу с последующим восстановлением. Ароматические соединения с сильнодезактивирующими заместителями, например, нитро- или циано- группами, также не ацилируются по Фриделю-Крафтсу.
Контрольные задачи
2. Изобразите диаграмму потенциальной энергии
для реакции электрофильного ароматического
замещения, в которой медленной стадией является
образование
-комплекса (например,
нитрование бензола борфторидом нитрония;
см. раздел IV.1.A).
3. Какой продукт преимущственно образуется при бромировании: а)пара -нитротолуола; б) мета -нитробензолсульфокислоты; в) орто -нитрофенола.
4. Адреналин (1-(3",4"-дигидроксифенил)-2-метиламиноэтанол)- первый гормон, выделенный из мозгового вещества надпочечников, в настоящее время синтезируюют в три стадии из пирокатехина. Напишите уравнение первой стадии этого синтеза - реакции ацилирования пирокатехина (1,2-дигидроксибензола) хлорангидридом хлоруксусной киcлоты и объясните механизм).
5. Одной их качественных реакций на белки является ксантопротеиновая реакция, указывающая на присутствие ароматических -аминокислот. Она заключается в обработке белка азотной кислотой при нагревании. Напишите уравнение ксантопротеиновой реакции с тирозином (см. раздел I), образовавшимся в результате гидролиза белка.
Больше трех нитрогрупп в молекулы бензола и толуола нитрованием ввести не удается.
Амино-2-нитротолуол л-Нитро-п-то- луидин сн Л ч/ 1 мн 0, и Толуол Нитрование - восстановление сернистым натром
М. С. Быховская описывает метод раздельного определения бензола и толуола. Нитрованием бензол переводят в динитробензол, а толуол- в тринитротолуол 77
При фракционировании жидкой части катализата было выделено 14,5 г углеводорода с т. кип. -ИГ (756 mai) n 1,4967 df 0,8665. Сопоставление констант этой фракции с литературными данными для толуола показывает, что она состоит из толуола.
Нитрованием этой фракции получен нитропродукт с т. пл. 69° С. Смешанная проба плавления с 2,4-динитротолуолом депрессии не дала.
Производство ксилила. Как мы видели из работ Мартинсена, при введении 2-й метильной группы в ядро быстроту реакции нитрации значительно увеличивается. Быстроту реакции нитрации ти-кси-лола в раз больше, чем толуола.
Поэтому и смеси для нитрования ксилола могут быть значительно слабее, чем для нитрования толуола. Этим же обусловлено то, что ксилол можно даже в заводских условиях нитровать в тринитроксилол в одну фазу , в то время как для толуола нитрование в одну фазу никогда и нигде не производилось, вследствие того, что это сопряжено с огромным расходом кислот, а потому неэкономично, не говоря о других трудностях а невыгодах нитрования толуола в тринитротолуол в одну фазу .
Образование значительных количеств жета-замещенных продуктов при алкилировании толуола и других монозамещенных бензолов можно объяснить высокой реакционной способностью атакующего реагента. Поскольку бромирование является примером достаточно мягкой реакции замещения, в данном случае сильно проявляются различия между бензолом и толуолом, а также между мета- и пара-положениями в толуоле.
Нитрование менее селективно, чем бромирование изопропилирование значительно менее селективно, чем нитрование. При алкилировании толуола образуется 30% лета-изомера. Более того, при этой реакции становятся незначительными различия между толуолом и бензолом.
Обзор реакций замещения в ароматическом ряду позволяет провести параллель между селективностью реакций с бензолом и толуолом, с одной стороны, и между мета- и пара-положениями в толуоле - с другой. В обоих случаях селективность уменьшается с увеличением реакционной сиособности атакующего агента 2. Данные табл. 4 иллюстрируют эти положения.70
При рассмотрении приведенных в литературе способов определения толуола представлялось возможным использовать фотометрический метод. Последний нашел широкое применение для определения малых количеств толуола, в частности в сточных водах и в воздухе промышленных предприятий. В основе многих вариантов колориметрических определений толуола - нитрование и последующее взаимодействие полученного нитросоединения со щелочью или аммиаком в различных растворителях ацетоне, спирте, метилэтилкетоне, бутаноле, спирто-эфирной смеси и других.
Зависимо от интенсивности охлаждения и перемешивания прилива-ние толуола продолжается 4-8 час. температуру нитрования поддерживают около 50° и по окончании приливания нагревают еще в течение. % часа при 80-90°. Более длительное и сильное нагревание нецелесообразно, так как после окончания приливания толуола нитрование
вряд ли продолжается.
Например при продолжении нагревания при 90° в течение 2 г час. температура затвердевания динитротолуола повысилась в одном случае с 33,7° только до 35,6°. По окончании нитрования содержимое аппарата охлаждают до температуры несколько более высокой, чем температура. плавления динитропродукта, и передавливают содержимое ии-тратора с помощью сжатого воздуха в одии из освинцованных сепараторов (1,2), смешивая реакционную смесь с таким количеством воды, чтобы разбавление достигало приблизительно 16%. При этом вначале происходит очень сильное выделение окислов азота, для отвода которых требуется достаточно мощная вытяжная труба. После отстаивания в течение нескольких часов при температуре несколько более высокой, чем температура плавления нитропродукта, кислота отделяется от продукта, образующего-верхний слой.377
Рубрики
Выберите рубрику 1. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА НЕФТИ, ПРИРОДНОГО ГАЗА 3. ОСНОВЫ РАЗРАБОТКИ НЕФТЯНЫХ МЕСТОРОЖДЕНИЙ И ЭКСПЛУАТА 3.1. Фонтанная эксплуатация нефтяных скважин 3.4. Эксплуатация скважин погружными электроцентробежны 3.6. Понятие о разработке нефтяных и газовых скважин 7. МЕТОДЫ ВОЗДЕЙСТВИЯ НА ПРИЗАБОЙНУЮ ЗОНУ ПЛАСТА ОСНОВНЫЕ УЗЛЫ ИСПЫТАТЕЛЯ ПЛАСТОВ ВИНТОВЫЕ ЗАБОЙНЫЕ ДВИГАТЕЛИ АВАРИЙНЫЕ И ОСОБЫЕ РЕЖИМЫ РАБОТЫ ЭЛЕКТРООБОРУДОВАНИЯ АГРЕГАТЫ ДЛЯ РЕМОНТА И БУРЕНИЯ СКВАЖИН АНАЛИЗ ПРИЧИН МАЛОДЕБИТНОСТИ СКВАЖИН АНАЛИЗ ТЕХНОЛОГИЙ КАПИТАЛЬНЫХ РЕМОНТОВ СКВАЖИН Арматура устьевая АСФАЛЬТОСМОЛО-ПАРАФИНОВЫЕ ОТЛОЖЕНИЯ Без рубрики БЕЗДЫМНОЕ СЖИГАНИЕ ГАЗА БЕСШТАНГОВЫЕ СКВАЖИННЫЕ НАСОСНЫЕ УСТАНОВКИ блогун БЛОКИ ЦИРКУЛЯЦИОННЫХ СИСТЕМ. борьба с гидратами БОРЬБА С ОТЛОЖЕНИЕМ ПАРАФИНА В ПОДЪЕМНЫХ ТРУБАХ бурение Бурение боковых стволов БУРЕНИЕ НАКЛОННО НАПРАВЛЕННЫХ И ГОРИЗОНТАЛЬНЫХ СКВАЖИН Бурение скважин БУРИЛЬНАЯ КОЛОННА БУРОВЫЕ АВТОМАТИЧЕСКИЕ СТАЦИОНАРНЫЕ КЛЮЧИ БУРОВЫЕ АГРЕГАТЫ И УСТАНОВКИ ДЛЯ ГЕОЛОГО-РАЗВЕДОЧНОГО БУРЕНИЯ БУРОВЫЕ ВЫШКИ БУРОВЫЕ НАСОСЫ БУРОВЫЕ НАСОСЫ БУРОВЫЕ РУКАВА БУРОВЫЕ УСТАНОВКИ В МНОГОЛЕТНЕМЕРЗЛЫХ ПОРОДАХ (ММП) ВЕНТИЛИ. ВИДЫ НЕОДНОРОДНОСТЕЙ СТРОЕНИЯ НЕФТЯНЫХ ЗАЛЕЖЕЙ Виды скважин ВИНТОВЫЕ ПОГРУЖНЫЕ НАСОСЫ С ПРИВОДОМ НА УСТЬЕ ВЛАГОСОДЕРЖАНИЕ И ГИДРАТЫ ПРИРОДНЫХ ГАЗОВ СОСТАВ ГИДРАТ Влияние различных факторов на характеристики ВЗД ВОПРОСЫ ОПТИМИЗАЦИИ РАБОТЫ СИСТЕМЫ ПЛАСТ — УЭЦН ВЫБОР ОБОРУДОВАНИЯ И РЕЖИМА РАБОТЫ УЭЦН ВЫБОР СТАНКА-КАЧАЛКИ Газлифтная установка ЛН Газлифтная эксплуатация нефтяных скважин Газлифтный способ добычи нефти ГАЗЫ НЕФТЯНЫХ И ГАЗОВЫХ МЕСТОРОЖДЕНИЙ И ИХ СВОЙСТВА ГИДРАТООБРАЗОВАНИЕ В ГАЗОКОНДЕНСАТНЫХ СКВАЖИНАХ ГИДРАТООБРАЗОВАНИЕ В СИСТЕМЕ СБОРА НЕФТИ гидрозащита погружного электродвигателя ГИДРОКЛЮЧ ГКШ-1500МТ гидропоршневой насос Глава 8. СРЕДСТВА И МЕТОДЫ ГРАДУИРОВКИ И ПОВЕРКИ РАСХОДОИЗМЕРИТЕЛЬНЫХ СИСТЕМ ГЛУБИННЫЕ НАСОСЫ Горизонтальное бурение ГОРНО-ГЕОЛОГИЧЕСКИЕ УСЛОВИЯ БУРЕНИЯ НЕФТЯНЫХ И ГАЗОВЫХ СКВАЖИН ГРАНУЛОМЕТРИЧЕСКИЙ (МЕХАНИЧЕСКИЙ) СОСТАВ ПОРОД ДАЛЬНИЙ ТРАНСПОРТ НЕФТИ И ГАЗА ДЕФОРМАЦИОННЫЕ МАНОМЕТРЫ Диафрагменные электронасосы ДИЗЕЛЬ-ГИДРАВЛИЧЕСКИЙ АГРЕГАТ САТ-450 ДИЗЕЛЬНЫЕ И ДИЗЕЛЬ-ГИДРАВЛИЧЕСКИЕ АГРЕГАТЫ ДИНАМОМЕТРИРОВАНИЕ УСТАНОВОК ДНУ С ЛМП КОНСТРУКЦИИ ОАО «ОРЕНБУРГНЕФТЬ» добыча нефти добыча нефти в осложненых условиях ДОБЫЧА НЕФТИ С ПРИМЕНЕНИЕМ ШСНУ ЖИДКОСТНЫЕ МАНОМЕТРЫ ЗАБОЙНЫЕ ДВИГАТЕЛИ Закачка растворов кислот в скважину ЗАПОРНАЯ АРМАТУРА. ЗАЩИТа НЕФТЕПРОМЫСЛОВОГО ОБОРУДОВАНИЯ ОТ КОРРОЗИИ ЗАЩИТА ОТ КОРРОЗИИ НЕФТЕПРОМЫСЛОВОГО ОБОРУДОВАНИЯ ИЗМЕНЕНИЕ КУРСА СТВОЛА СКВАЖИНЫ измерение давления, расхода, жидкости, газа и пара ИЗМЕРЕНИЕ КОЛИЧЕСТВА ЖИДКОСТЕЙ И ГАЗОВ ИЗМЕРЕНИЕ РАСХОДА ЖИДКОСТЕЙ, ГАЗОВ И ПАРОВ ИЗМЕРЕНИЕ УРОВНЯ ЖИДКОСТЕЙ ИЗМЕРЕНИЯ ПРОДУКЦИИ МАЛОДЕБИТНЫХ ИНФОРМАЦИОННЫЕ ТЕХНОЛОГИИ В НЕФТЕГАЗОДОБЫЧЕ ИСПЫТАНИЕ СКВАЖИННЫХ ЭЛЕКТРОНАГРЕВАТЕЛЕЙ Исследование глубинно-насосных скважин ИССЛЕДОВАНИЕ ЭФФЕКТИВНОСТИ кабель УЭЦН капитальный ремонт скважин Комплекс оборудования типа КОС и КОС1 КОНСТРУКЦИЯ ВИНТОВОГО ШТАНГОВОГО НАСОСА КОНСТРУКЦИЯ КЛАПАННОГО УЗЛА коррозия Краны. КРЕПЛЕНИЕ СКВАЖИН КТППН МАНИФОЛЬДЫ Маятниковая компоновка Меры безопасности при приготовлении растворов кислоты МЕТОДИКА РАСЧЕТА БУРИЛЬНЫХ КОЛОНН МЕТОДЫ БОРЬБЫ С ОТЛОЖЕНИЯМИ ПАРАФИНА В ФОНТАННЫХ СКВАЖИНАХ Методы воздействия на призабойную зону для увеличения нефтеотдачи пластов МЕТОДЫ И СРЕДСТВА ИЗМЕРЕНИЯ УРОВНЯ ЖИДКОСТЕЙ Методы изучения разрезов скважин. МЕТОДЫ КОСВЕННЫХ ИЗМЕРЕНИЙ ДАВЛЕНИЯ МЕТОДЫ УДАЛЕНИЯ СОЛЕЙ МЕХАНИЗМЫ ПЕРЕДВИЖЕНИЯ И ВЫРАВНИВАНИЯ БУРОВЫХ УСТАНОВОК МЕХАНИЗМЫ ПЕРЕМЕЩЕНИЯ И ВЫРАВНИВАНИЯ МЕХАНИЗМЫ ПРИ СПУСКО-ПОДЪЕМНЫХ ОПЕРАЦИЙ ПРИ БУРЕНИИ НАГРУЗКИ, ДЕЙСТВУЮЩИЕ НА УСТАНОВКУ Наземное оборудование Насосная эксплуатация скважин НАСОСНО-КОМПРЕССОРНЫЕ ТРУБЫ неоднородный пласт Нефть и нефтепродукты Новости портала НОВЫЕ ТЕХНОЛОГИЧЕСКИЕ И ТЕХНИЧЕСКИЕ ОБЕСПЕЧЕНИЕ ЭКОЛОГИЧЕСКОЙ БЕЗОПАСНОСТИ ПРОЦЕССОВ ДОБЫЧИ ОБОРУДОВАНИЕ ГАЗЛИФТНЫХ СКВАЖИН ОБОРУДОВАНИЕ ДЛЯ МЕХАНИЗАЦИИ СПУСКО-ПОДЪЕМНЫХ ОПЕРАЦИЙ Оборудование для нефти и газа ОБОРУДОВАНИЕ ДЛЯ ОДНОВРЕМЕННОЙ РАЗДЕЛЬНОЙ ЭКСПЛУАТАЦ ОБОРУДОВАНИЕ ДЛЯ ПРЕДУСМОТРЕНИЯ ОТКРЫТЫХ ФОНТАНОВ ОБОРУДОВАНИЕ ОБЩЕГО НАЗНАЧЕНИЯ Оборудование ствола скважины, законченной бурением ОБОРУДОВАНИЕ УСТЬЯ КОМПРЕССОРНЫХ СКВАЖИН ОБОРУДОВАНИЕ УСТЬЯ СКВАЖИНЫ Оборудование устья скважины для эксплуатации УЭЦН ОБОРУДОВАНИЕ ФОНТАННЫХ СКВАЖИН ОБОРУДОВАНИЕ ФОНТАННЫХ СКВАЖИН обработка призабойной зоны ОБРАЗОВАНИЕ ГИДРАТОВ И МЕТОДЫ БОРЬБЫ С НИМИ ОБРАЗОВАНИЕ КРИСТАЛЛОГИДРАТОВ В НЕФТЯНЫХ СКВАЖИНАХ ОБЩИЕ ПОНЯТИЯ О ПОДЗЕМНОМ И КАПИТАЛЬНОМ РЕМОНТЕ ОБЩИЕ ПОНЯТИЯ О СТРОИТЕЛЬСТВЕ СКВАЖИН ОГРАНИЧЕНИЕ ПРИТОКА ПЛАСТОВЫХ ВОД Опасные и вредные физические факторы ОПРЕДЕЛЕНИЕ ДАВЛЕНИЯ НА ВЫХОДЕ НАСОСА ОПРОБОВАНИЕ ПЕРСПЕКТИВНЫХ ГОРИЗОНТОВ ОПТИМИЗАЦИЯ РЕЖИМА РАБОТЫ ШСНУ ОПЫТ ЭКСПЛУАТАЦИИ ДНУ С ГИБКИМ ТЯГОВЫМ ЭЛЕМЕНТОМ ОСВОЕНИЕ И ИСПЫТАНИЕ СКВАЖИН ОСВОЕНИЕ И ПУСК В РАБОТУ ФОНТАННЫХ СКВАЖИН ОСЛОЖНЕНИЯ В ПРОЦЕССЕ УГЛУБЛЕНИЯ СКВАЖИНЫ ОСНОВНЫЕ ПОНЯТИЯ И ПОЛОЖЕНИЯ ОСНОВНЫЕ ПОНЯТИЯ И ПОЛОЖЕНИЯ ОСНОВНЫЕ СВЕДЕНИЯ О НЕФТЯНЫХ, ГАЗОВЫХ И ГАЗОКОНДЕНСАТНЫ ОСНОВЫ ГИДРАВЛИЧЕСКИХ РАСЧЕТОВ В БУРЕНИИ ОСНОВЫ НЕФТЕГАЗОДОБЫЧИ ОСНОВЫ ПРОЕКТИРОВАНИЯ НАПРАВЛЕННЫХ СКВАЖИН ОСНОВЫ ПРОМЫШЛЕННОЙ БЕЗОПАСНОСТИ ОЧИСТКА БУРЯЩЕЙСЯ СКВАЖИНЫ ОТ ШЛАМА ОЧИСТКА ПОПУТНЫХ ГАЗОВ пайка и наплавка ПАКЕР ГИДРОМЕХАНИЧЕСКИЙ ДВУХМАНЖЕТНЫЙ ПГМД1 ПАКЕРЫ ГИДРОМЕХАНИЧЕСКИЕ, ГИДРАВЛИЧЕСКИЕ И МЕХАНИЧЕСКИЕ ПАКЕРЫ ДЛЯ ИСПЫТАНИЯ КОЛОНН ПАКЕРЫ РЕЗИНОВО-МЕТАЛЛИЧЕСКОГО ПЕРЕКРЫТИЯ ПРМП-1 ПАКЕРЫ И ЯКОРИ ПАРАМЕТРЫ И КОМПЛЕКТНОСТЬ ЦИРКУЛЯЦИОННЫХ СИСТЕМ Параметры талевых блоков для работы с АСП ПЕРВИЧНОЕ ВСКРЫТИЕ ПРОДУКТИВНЫХ ПЛАСТОВ ПЕРВИЧНЫЕ СПОСОБЫ ЦЕМЕНТИРОВАНИЯ ПЕРЕДВИЖНЫЕ НАСОСНЫЕ УСТАНОВКИ И АГРЕГАТЫ ПЕРЕРАБОТКА ЛОВУШЕЧНЫХ НЕФТЕЙ (НЕФТЕШЛАМОВ) ПЕРИОДИЧЕСКИЙ ГАЗЛИФТ ПЕРСПЕКТИВЫ ИСПОЛЬЗОВАНИЯ ДНУ ПОВЫШЕНИЕ ЭФФЕКТИВНОСТИ ПОВЫШЕНИЕ ЭФФЕКТИВНОСТИ РАБОТЫ ШСНУ Погружение насосов под динамический уровень Подземное оборудование фонтанных скважин ПОДЪЕМ ВЯЗКОЙ ЖИДКОСТИ ПО ЗАТРУБНОМУ ПРОСТРАНСТВУ СКВАЖИНЫ ПОРОДОРАЗРУШАЮЩИЕ ИНСТРУМЕНТЫ ПОРШНЕВЫЕ МАНОМЕТРЫ Потери давления при движении жидкости по нкт Правила безопасности при эксплуатации скважин Правила ведения ремонтных работ в скважинах РД 153-39-023-97 ПРЕДУПРЕЖДЕНИЕ ОБРАЗОВАНИЯ СОЛЕЙ ПРЕДУПРЕЖДЕНИЯ ОБРАЗОВАНИЯ АСПО ПРЕДУПРЕЖДЕНИЯ ОБРАЗОВАНИЯ АСПО при работе ШГН ПРЕИМУЩЕСТВА ДЛИННОХОДОВЫХ Приготовление растворов кислот. ПРИГОТОВЛЕНИЕ, ОЧИСТКА БУРОВЫХ РАСТВОРОВ ПРИМЕНЕНИЕ СТРУЙНЫХ КОМПРЕССОРОВ ДЛЯ УТИЛИЗАЦИИ ПРИМЕНЕНИЕ УЭЦН В СКВАЖИНАХ ОАО «ОРЕНБУРГНЕФТЬ» ПРИНЦИП ДЕЙСТВИЯ И ОСОБЕННОСТИ КОНСТРУКЦИИ ДНУ С ЛМП ПРИЧИНЫ И АНАЛИЗ АВАРИЙ ПРОГНОЗИРОВАНИЕ ОТЛОЖЕНИЯ НОС ПРИ ДОБЫЧЕ НЕФТИ ПРОЕКТИРОВАНИЕ ТРАЕКТОРИИ НАПРАВЛЕННЫХ СКВАЖИН ПРОЕКТИРОВАНИЕ, ОБУСТРОЙСТВО И АНАЛИЗ РАЗРАБОТКИ УГЛЕВОДОРОДНЫХ МЕСТОРОЖДЕНИЙ Производительность насоса ПРОМЫВКА СКВАЖИН И БУРОВЫЕ РАСТВОРЫ ПРОМЫСЛОВЫЕ ИССЛЕДОВАНИЯ ПРОМЫСЛОВЫЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ ЗОН ОБРАЗОВАНИЯ НОС ПРОМЫСЛОВЫЙ СБОР И ПОДГОТОВКА НЕФТИ, ГАЗА И ВОДЫ ПРОТИВОВЫБРОСОВОЕ ОБОРУДОВАНИЕ ПУТИ ПОВЫШЕНИЯ ЭФФЕКТИВНОСТИ ЭКСПЛУАТАЦИИ СКВАЖИН РАЗМЕЩЕНИЕ ЭКСПЛУАТАЦИОННЫХ И НАГНЕТАТЕЛЬНЫХ СКВАЖИН НА Разное РАЗРУШЕНИЕ ГОРНЫХ ПОРОД РАСПРЕДЕЛЕНИЕ ОБРЫВОВ ПО ДЛИНЕ КОЛОННЫ ШТАНГ РАСЧЕТ ДНУ РАСЧЕТ ПРОИЗВОДИТЕЛЬНОСТИ ДНУ Регулирование свойств цементного раствора и камня с помощью реагентов Режимы добывающих и нагнетательных скважин. РЕЗЕРВЫ СНИЖЕНИЯ ЭНЕРГОПОТРЕБЛЕНИЯ ПРИ ЭКСПЛУАТАЦИ РЕМОНТЫ ПО ЭКОЛОГИЧЕСКОМУ ОЗДОРОВЛЕНИЮ ФОНДА СКВАЖИН РОЛЬ ФОНТАННЫХ ТРУБ САМОХОДНЫЕ УСТАНОВКИ С ПОДВИЖНЫМ… СЕТКА РАЗМЕЩЕНИЯ СКВАЖИН СИСТЕМЫ УЛАВЛИВАНИЯ ЛЕГКИХ УГЛЕВОДОРОДОВ Скважинные уплотнители (пакеры) Скважинные центробежные насосы для добычи нефти СОСТАВ И НЕКОТОРЫЕ СВОЙСТВА ВОД НЕФТЯНЫХ И ГАЗОВЫХ МЕСТ СПЕЦИАЛЬНЫЙ НЕВСТАВНОЙ ШТАНГОВЫЙ НАСОС СПОСОБЫ ДОБЫЧИ НЕФТИ, ПРИМЕНЯЕМЫЕ НА МЕСТОРОЖДЕНИЯХ ОАО СПОСОБЫ ОЦЕНКИ СОСТОЯНИЯ ПЗП СРАВНИТЕЛЬНЫЕ ИСПЫТАНИЯ НАСОСНЫХ УСТАНОВОК СРЕДСТВА И МЕТОДЫ ПОВЕРКИ СЧЕТЧИКОВ КОЛИЧЕСТВА ГАЗОВ СРЕДСТВА И МЕТОДЫ ПОВЕРКИ СЧЕТЧИКОВ КОЛИЧЕСТВА ЖИДКОСТЕЙ СТАДИИ РАЗРАБОТКИ МЕСТОРОЖДЕНИЙ Станки-качалки Струйные насосы струйный насос СЧЕТЧИКИ КОЛИЧЕСТВА ГАЗОВ СЧЕТЧИКИ КОЛИЧЕСТВА ЖИДКОСТЕЙ ТАЛЕВЫЕ МЕХАНИЗМЫ ТЕМПЕРАТУРА И ДАВЛЕНИЕ В ГОРНЫХ ПОРОДАХ И СКВАЖИНАХ Теоретические основы безопасности ТЕХНИКА ИЗМЕРЕНИЯ РАСХОДА Техническая физика ТРАЕКТОРИЮ ПЕРЕМЕЩЕНИЯ ЗАБОЯ СКВАЖИНЫ Трубы УКАЗАНИЯ ПО РАСЧЕТУ ТОКОВ КОРОТКОГО ЗАМЫКАНИЯ УСЛОВИЯ ПРИТОКА ЖИДКОСТИ И ГАЗА В СКВАЖИНЫ Установки гидропоршневых насосов для добычи нефти Установки погружных винтовых электронасосов Установки погружных диафрагменных электронасосов Устьевое оборудование УТЯЖЕЛЕННЫЕ БУРИЛЬНЫЕ ТРУБЫ УЭЦН уэцн полностью ФАКТОРЫ, ВЛИЯЮЩИЕ НА ИНТЕНСИВНОСТЬ ОБРАЗОВАНИЯ АСПО Физико-механические свойства пород-коллекторов ФИЗИЧЕСКАЯ ХАРАКТЕРИСТИКА ГАЗОВ НЕФТЯНЫХ И ГАЗОВЫХ МЕСТ ФИЛЬТРЫ Фонтанный способ добычи нефти ЦЕМЕНТИРОВАНИЕ СКВАЖИН ЦИРКУЛЯЦИОННЫЕ СИСТЕМЫ БУРОВЫХ УСТАНОВОК Шлакопесчаные цементы Шлакопесчаные цементы совместного помола Штанги насосные (ШН) ШТАНГОВЫЕ НАСОСНЫЕ УСТАНОВКИ (ШСНУ) ШТАНГОВЫЕ НАСОСЫ ДЛЯ ПОДЪЕМА ВЯЗКОЙ НЕФТИ ШТАНГОВЫЕ СКВАЖИННЫЕ НАСОСЫ Штанговые скважинные насосы ШСН ЭКСПЛУАТАЦИЯ ГАЗОВЫХ СКВАЖИН эксплуатация малодебитных скважин ЭКСПЛУАТАЦИЯ МАЛОДЕБИТНЫХ СКВАЖИН НА НЕПРЕРЫВНОМ РЕЖИМЕ ЭКСПЛУАТАЦИЯ ОБВОДНЕННЫХ ПАРАФИНСОДЕРЖАЩИХ СКВАЖИН ЭКСПЛУАТАЦИЯ СКВАЖИН ЭКСПЛУАТАЦИЯ СКВАЖИН УЭЦН ЭЛЕКТРОДЕГИДРАТОР. ЭЛЕКТРОДИАФРАГМЕННЫЙ НАСОС энергосбережение скважинного электронасосного агрегата ЯКОРИ1 .. 40 > .. >> Следующая
Неочищенный тротил можно применять только для изготовления взрывчатых смесей, предназначенных к быстрому употреблению, например для взрывных работ. Тротил же, идущий для снаряжения боеприпасов, которые подлежат длительному хранению, должен быть обязательно подвергнут очистке.
ТЕХНОЛОГИЯ ПОЛУЧЕНИЯ ТРОТИЛА
Закономерности процесса нитрования
Нитрование толуола до мононитротолуола. При нитровании толуола до мононитротолуола получаются три изомера: орто-, пара- и мета, с преимущественным преобладанием орто-то-мера. .мета-Изомер образуется в относительно небольшом количестве, но так как при его дальнейшем нитровании получаются несимметричные трйнитротолуолы, то образование его нежелательно.
Основные работы по исследованию закономерностей реакции нитрования толуола до мононитротолуола были направлены главным образом на выявление условий наименьшего выхода лета-изомера. Когда непрерывные процессы нитрования стали доминирующими и назрел вопрос о рациональных конструкциях нитраторов, на"чалось изучение кинетики нитрования, в основном в гетеро-
Рис. 17. Растворимость толуола в серной кислоте различной концентрации при 55 °С.
65 70 75 80 85 90 Концентрация HzS047%
95
генной среде, что соответствует промышленным условиям. При этом исследовали растворимость толуола в серно-азотных кислотных смесях, распределение компонентов между слоями, влияние перемешивания и соотношения объемов органического и кислотного слоев на скорость реакции. Растворимость толуола в серной кислоте растет с повышением концентрации кислоты; до концентрации кислоты 80% растворимость толуола очень низка (рис. 17).
Скорость нитрования толуола в гетерогенных условиях резко зависит от интенсивности перемешивания и модуля ванны (отношение объемов минерального и органического слоев) (рис. 18).
Коэффициент распределения азотной кислоты между толуоль-ным и серно-кислотным слоями равен 0,066. Это-указывает на то, что азотная кислота при гетерогенном нитровании толуола лишь в незначительной степени переходит в органический слой и поэтому доля протекающей там реакции практически равна нулю.
Низкая растворимость толуола в серной кислоте умеренных концентраций, отсутствие перехода азотной кислоты в органический слой, а также резкая зависимость скорости реакции нитрования толуола от интенсивности перемешивания и объемной доли минерального слоя позволяют предположить, что реакция нитрования толуола в гетерогенных условиях протекает на поверхности раздела слоев. Скорость реакции в этом случае зависит от концентрации реагирующих компонентов^ на этой поверхности, которая в свою очередь определяется скоростью диффузии реагирующих компонентов из глубины слоя к поверхности раздела и скоростью отхода от нее продуктов реакции. Все это, а также состояние реагирующих компонентов зависит от температуры (рис. 19, с), концентрации кислотной смеси (рис. 19, б) и интенсивности перемешивания (рис. 18), причем скорость нитрования толуола в гетерогенных условиях ниже, чем в гомогенных (рис. 19,6).
Образование 5-6% л-нитротолуола при нитровании толуола в дальнейшем приводит к образованию 5-6% несимметричных тринитротолуолов, загрязняющих тротил. Температура затвердева-
Рис. 18. Влияние интенсивности перемешивания (а) и модуля ианны (6") на степень нитрования толуола.
ния тротила, содержащего несимметричные изомеры, снижается по следующей зависимости:
Г3 = (80,80 - 0.465С)
где С - содержание л-нитротолуола в исходном мононитротолуоле, %.
На изомерный состав мононитротолуола значительное влияние оказывает температура нитрования толуола (см. стр. 85). Исследование скорости нитрования толуола при 0 и 30 °С и определение изомерного состава позволило рассчитать в уравнении Аррениуса (см. стр. 54) коэффициент В для вступления нитрогруппы в различные положения относительно СН3-группы: В0 = 2,90 Вя, Вп = 2,70 Вм\ энергия активации для соответствующих положений равна: Е„ - Е0 = 3,83 кДж/(моль-°С), Е„ - ?„ = 4,61 кДж/(моль-°С). Из этих данных может быть сформулировано следующее правило" понижение температуры нитрования способствует увеличению выхода п-нитротолуола и уменьшению выхода о- и м-нитротолуолов.
Значительное влияние на выход л-нитротолуола оказывает фактор нитрующей активности кислотной смеси. Повышение Ф с 68 до 82% при нитровании толуола серно-азотными кислотными смесями при 55 °С снижает выход лета-изомера в 2,4 раза. На рис. 20 показано влияние температуры и фактора нитрующей активности кислотной смеси на выход л-нитротолуола.
Применение для нитрования толуола разбавленной азотной кислоты (70%-ной) приводит к образованию продуктов окисления, главным образом бензойной кислоты. Применение еще более разбавленной кислоты (32%-ной) при повышенной температуре (105 °С) вызывает нитрование боковой цепи, образуется фенил-нитрометан, который получается также и при нитровании толуола двуокисью азота.
-l-I-а,; ,„i, I_I I-1_¦ \ ¦_I
25 зо 40 60 № 70 во 64 ее ее ю 72 74
Твмперашура,°С Ф
Рис. 19. Зависимость скорости нитрования толуола от температуры (а) и фактора нитрующей активности Ф (6*):
Нитрование бензола может быть проведено с небольшими количествами исходных веществ, без выделения чистого продукта. Для получения нитробензола по уравнению:
С 6 Н 6 + HNO 3 à C 6 H 5 NO 2 + Н 2 О
необходима концентрированная азотная кислота (уд. вес. 1,4). Реакционная смесь при этом не должна нагреваться выше 50-60"С. При применении разбавленной кислоты реакция нитрования не идет; при повышении температуры начинается заметное образование динитробензола.
Из уравнения следует, что для реакции необходимы эквимолекулярные количества исходных веществ. Однако в таком случае реакция не дойдет до конца, так как выделяющаяся вода будет разбавлять азотную кислоту, и она потеряет нитрующее свойство. Следовательно, чтобы довести реакцию до конца, надо взять больше азотной кислоты, чем следует по теории. Но, чтобы реакция при этом не стала слишком бурной, азотную кислоту нужно растворить в концентрированной серной кислоте, которая не лишает азотную кислоту нитрующего действия и связывает выделяющуюся при реакции воду.
Чтобы предупредить возможность повышения температуры при реакции, не смешивают сразу все вещества, а к смеси кислот постепенно добавляют бензол. В небольшую колбочку наливают 8 мл концентрированной серной кислоты и 5 мл концентрированной азотной кислоты. Охлаждают смесь в струе воды. Затем к охлажденной смеси прибавляют небольшими порциями 4 мл бензола, постоянно встряхивая колбочку, чтобы достичь большего смешения нерастворяющихся друг в друге жидкостей (смесь кислот составляет нижний слой, бензол - верхний слой). После приливания всего бензола для достижения полноты реакции колбу закрывают пробкой с вертикальной трубкой (пары бензола летучи) и нагревают на предварительно нагретой до 60°С водяной бане
Время от времени колбу встряхивают, чтобы жидкости лучше перемешивались.
Продолжительность нагревания может определяться не столько необходимостью достижения полноты реакции, сколько наличием времени на уроке. При работе в кружке нагревание следует продолжать минут 30-40. Па уроке же удается продемонстрировать образование нитробензола после нагревания в течение 10 мин и даже вовсе без дополнительного нагревания, если реакция хорошо шла при приливании бензола к смеси кислот.
Нитробензол располагается слоем поверх смеси кислот. Выливают содержимое колбы в стакан с большим количеством воды. При этом кислоты растворяются в воде, нитробензол же собирается на дне стакана в виде тяжелой желтоватой жидкости. Если позволяет время, сливают часть жидкости с нитробензола и отделяют его с помощью делительной воронки.
При получении значительных количеств нитробензола и необходимости его очистки, нитробензол промывают водой, разбавленным (5-процентным) раствором щелочи, затем снова водой, разделяя всякий раз жидкости с помощью делительной воронки. После этого обезвоживают нитробензол, нагревая его с гранулированным хлоридом кальция, пока жидкость не станет прозрачной. Нагревание при этом необходимо, чтобы понизить вязкость нитробензола и достичь таким образом более полного контакта его с хлоридом кальция. Наконец, нитробензол может быть перегнан из небольшой колбочки с воздушным холодильником при температуре 204-207°С. Для того чтобы избежать разложения остатков динитробензола, не рекомендуется проводить перегонку досуха.



